全基因组测序分为重测序(Whole Genome Resequencing) 和从头测序(Genome De-novo Sequencing)。重测序是对已有参考基因组物种的不同个体进行全基因组测序,并在此基础上对个体或群体进行差异性分析,在人类疾病和动植物育种研究中被广泛应用。
简介
人类已知疾病中约有8,000 多种与基因有关,利用基因组重测序可以辅助研究者发现与人类疾病相关的单核苷酸位点(SNV/SNP)、拷贝数变异(CNV)、插入/ 缺失(InDel)变异和结构变异(SV)等,从而进行疾病的致病或易感基因筛找、发病及遗传机制研究、药物靶点确认,以及推断种群迁移和进化等。在动植物育种中,则可以对动植物的遗传进化分析、性状候选基因等提供重要参考信息。
技术路线
技术优势

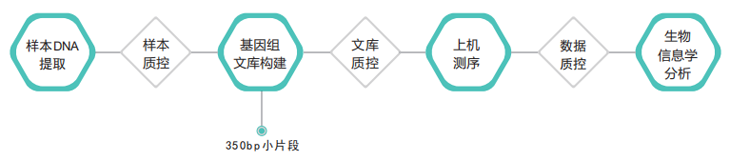
测序策略

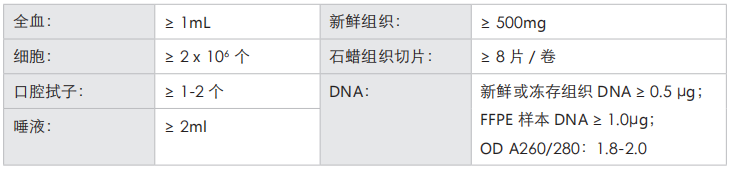
收样要求
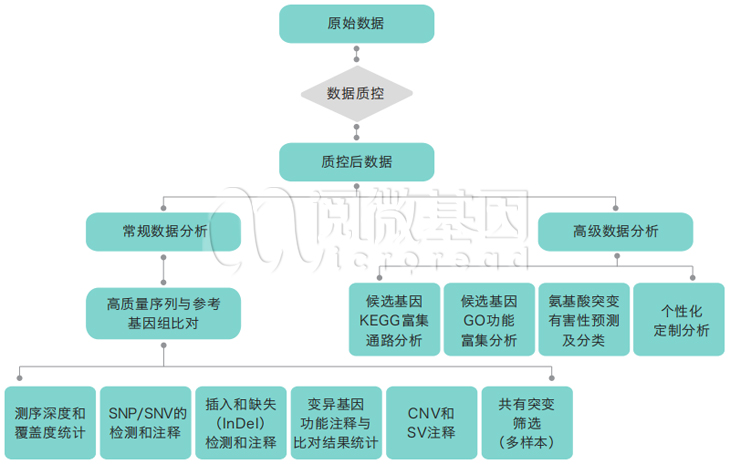
数据分析