



你是准备好做父母了,但你问过基因的意见了吗?
【2018-11-20】
双11已经过啦!单身的小伙伴,你们找到要携手终生的TA了吗?合适的伴侣不仅仅要彼此欣赏,基因上的“门当户对”对下一代也是万分关键的,因为基因上的优良结合,可以扬长避短,降低出生缺陷,孕育出健康的宝宝。
你身边是否有这样的场景:几个月的婴儿,吞咽困难、四肢肌张力减退,甚至呼吸衰竭而死亡;几岁的儿童,脊柱侧弯、经常骨折,始终无法独立行走,但家长却都身体健全。这就是常染色体隐性遗传病SMA(脊髓性肌萎缩症)的典型临床症状。
SMA是一种常见的神经肌肉病,患者脊髓前角运动神经元受到侵害,表现为进行性、对称性肌肉萎缩及肌肉无力。根据发病年龄、病变程度可将本病分为4型,SMA是导致婴幼儿死亡的头号遗传性疾病,在人群中的携带率为1/35-1/50[1]。
运动神经元存活(Survival Motor Neuron, SMN)基因是SMA的主要致病基因,定位于染色体5q11.2-q13.3区域,该基因具有两个高度同源的拷贝SMN1和SMN2。SMN1表达全长的有功能的SMN蛋白,SMN2由于第7外显子中c.840位点的C>T的碱基差异,表达的蛋白大部分跳跃了第7外显子,只产生少量全长蛋白,仅能在SMA患儿缺失SMN1基因时,起剂量补偿作用,因而,SMN1基因缺失或突变是SMA的主要病因,约95%的SMA患者存在SMN1基因第七外显子纯合缺失[2]。
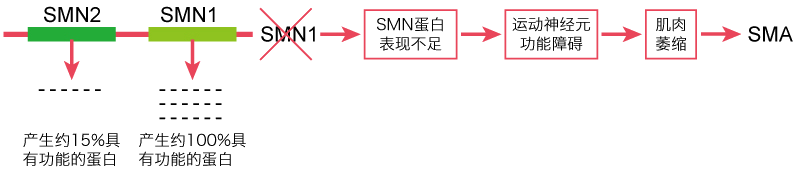
SMA属于携带率高、发病率高、易致残致死的一类隐性遗传疾病。研究表明,98%的SMA患儿,其父母都是携带者;只有2%的患儿由新发突变导致。
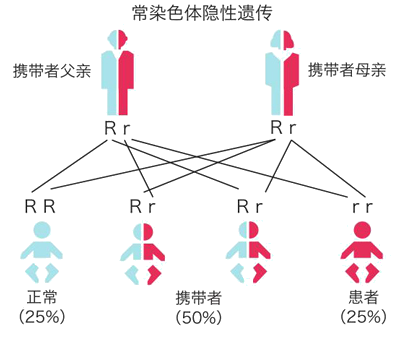
假如父母都是SMA携带者,那么生育的孩子有50%的可能会携带SMA致病基因,还有25%的可能会是SMA患儿。
目前对于SMA的治疗,上市药物Spinraza通过改变SMN2前体mRNA的剪接以增加全长SMN蛋白生成,来达到治疗SMA病的效果,但治疗费用昂贵。
如果新生儿为SMA患者,不管是患者本人还是患者的家庭,心理和经济上的负担都是不小的。所以对于SMA,预防的重要性并不比唐氏综合症低,通过婚检、孕前筛查、产前诊断,可以有效地避免SMA患儿的出生。
早在2008年,美国医学遗传学会(ACMG)建议对SMA泛族裔筛查,即不分区域、种族,对所有孕龄人群无差别实施SMN1携带者基因筛查;对高风险胎儿实施产前诊断,从而减少SMA患儿的出生。今年5月11日,我国已将SMA收录到第一批罕见病目录。
SMA基因筛查流程及干预方案:
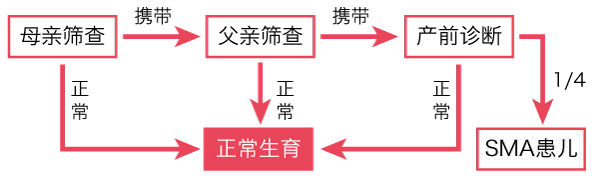
阅微基因SMA检测,仅需要2ml外周血,可同时检测SMN1/2基因拷贝数,有效区分正常人、携带者和患者。夫妻双方通过SMA基因检测后,若结果显示都是携带者,则需要继续做胎儿产前诊断(25%的概率为SMA患儿)。除用于SMA携带者筛查之外,SMA检测还可以用于SMA疾病辅助诊断。不管是否生育过SMA患儿,都建议进行SMA携带者筛查。所以啦,在你们准备好做父母之前,先做个检测才放心呀!
参考文献:
[1] Wilson, R. B., et al. (2008). Carrier frequency of spinal muscular atrophy. The Lancet. DOI:10.1016/S0140-6736(08)61646-1
[2] Parsons,D. W., et al. (1998). Intragenic telSMN Mutations: Frequency, Distribution, Evidence of a Founder Effect, and Modification of the Spinal Muscular Atrophy Phenotype by cenSMN Copy Number. American Journal of Human Genetics. DOI:10.1086/302160

你身边是否有这样的场景:几个月的婴儿,吞咽困难、四肢肌张力减退,甚至呼吸衰竭而死亡;几岁的儿童,脊柱侧弯、经常骨折,始终无法独立行走,但家长却都身体健全。这就是常染色体隐性遗传病SMA(脊髓性肌萎缩症)的典型临床症状。
SMA是什么?
SMA是一种常见的神经肌肉病,患者脊髓前角运动神经元受到侵害,表现为进行性、对称性肌肉萎缩及肌肉无力。根据发病年龄、病变程度可将本病分为4型,SMA是导致婴幼儿死亡的头号遗传性疾病,在人群中的携带率为1/35-1/50[1]。
运动神经元存活(Survival Motor Neuron, SMN)基因是SMA的主要致病基因,定位于染色体5q11.2-q13.3区域,该基因具有两个高度同源的拷贝SMN1和SMN2。SMN1表达全长的有功能的SMN蛋白,SMN2由于第7外显子中c.840位点的C>T的碱基差异,表达的蛋白大部分跳跃了第7外显子,只产生少量全长蛋白,仅能在SMA患儿缺失SMN1基因时,起剂量补偿作用,因而,SMN1基因缺失或突变是SMA的主要病因,约95%的SMA患者存在SMN1基因第七外显子纯合缺失[2]。
如需详细了解SMA发病机理,
请点击——《一场醒不来的噩梦-SMA》
为什么推荐孕龄人群进行SMA基因检测
假如父母都是SMA携带者,那么生育的孩子有50%的可能会携带SMA致病基因,还有25%的可能会是SMA患儿。
现状如何?
目前对于SMA的治疗,上市药物Spinraza通过改变SMN2前体mRNA的剪接以增加全长SMN蛋白生成,来达到治疗SMA病的效果,但治疗费用昂贵。
如果新生儿为SMA患者,不管是患者本人还是患者的家庭,心理和经济上的负担都是不小的。所以对于SMA,预防的重要性并不比唐氏综合症低,通过婚检、孕前筛查、产前诊断,可以有效地避免SMA患儿的出生。
早在2008年,美国医学遗传学会(ACMG)建议对SMA泛族裔筛查,即不分区域、种族,对所有孕龄人群无差别实施SMN1携带者基因筛查;对高风险胎儿实施产前诊断,从而减少SMA患儿的出生。今年5月11日,我国已将SMA收录到第一批罕见病目录。
SMA基因筛查流程及干预方案:
阅微基因SMA检测,仅需要2ml外周血,可同时检测SMN1/2基因拷贝数,有效区分正常人、携带者和患者。夫妻双方通过SMA基因检测后,若结果显示都是携带者,则需要继续做胎儿产前诊断(25%的概率为SMA患儿)。除用于SMA携带者筛查之外,SMA检测还可以用于SMA疾病辅助诊断。不管是否生育过SMA患儿,都建议进行SMA携带者筛查。所以啦,在你们准备好做父母之前,先做个检测才放心呀!
参考文献:
[1] Wilson, R. B., et al. (2008). Carrier frequency of spinal muscular atrophy. The Lancet. DOI:10.1016/S0140-6736(08)61646-1
[2] Parsons,D. W., et al. (1998). Intragenic telSMN Mutations: Frequency, Distribution, Evidence of a Founder Effect, and Modification of the Spinal Muscular Atrophy Phenotype by cenSMN Copy Number. American Journal of Human Genetics. DOI:10.1086/302160



